






医疗器械生物学评价依据与流程
医疗器械的生物学评价依据通常会考虑如下几个方面;
1)原材料组成;
2)添加剂、工艺污染物及残留物;
3)可沥滤物;
4)降解产物;
5)其他组件及其在终产品中的相互作用;
6)终产品的性能与特点;
7)终产品的物理特性等。
此外,还需根据具体产品的特定风险进行分析和评价。如植入类产品的局部作用、可吸收产品的降解测试、纳米材料器械应参考ISO/TR 10993-22等。
医疗器械的生物学评价流程
考虑器械潜在的生物学风险,并不意味着要针对所有风险点都开展生物学测试,还可通过“评价”的方式开展物理/化学表征及毒理学风险评估、基于已有的临床应用历史和人体接触数据对器械的生物学风险进行评估。在近年来更新的ISO 10993-1:2018和FDA年发布的关于ISO 10993-1的应用指南中,均强调了通过化学表征测试和毒理学风险评估(ISO 10993-18和ISO 10993-17)进行生物学评价的思路,不仅可以豁免不必要的生物相容性测试及避免人力、物力和动物资源的浪费,还可以基于已有的研究数据更加充分地评估器械(尤其是持久性植入的高风险类器械)中潜在的生物安全性风险(如慢性毒性、致癌性和生殖毒性等),从而优化生物学测试方案,最终达到器械安全性评价的目的。
具体评价流程如图1所示:

医疗器械材料组成和生产工艺中引入化学物质的定性和定量信息是进行医疗器械生物学评价和毒理学风险评估的重要信息输入,对于原材料成分和配方信息的收集和生成有助于辅助可浸提物或可沥滤物研究来鉴定器械材料迁出的化学物质。这些信息可以辅助毒理学家更好的开展毒理学风险评估进而指导生物相容性试验,最终评价医疗器械及其材料相关的潜在安全风险。
ISO10993-18:2020的更新,重点增加了可浸提物和可沥滤物研究相关的内容,例如标准中强调了分析阈值(AET)的重要性, 要确保分析方法的定量限或检出限满足分析阈值的要求。因为不同产品的接触时间和类型不同,且表面积的差异,导致分析阈值的限度差异很大且限度很低,开发合适的分析方法以满足全球监管的要求成为化学表征领域的一大难点。
其次,可浸提物和可沥滤物的研究的本质是未知物的发现以及鉴定,因此医疗器械相关未知物的鉴定也是化学表征领域的又一大难点,选择专业有经验的研究机构会帮助企业完善申报资料。ISO 10993-18: 2020的更新已经改变了医疗器械的测试和申报时间表,预计正在更新状态之中的ISO 10993-17(预计在2021-2022年发布)将在制定器械申报计划时在生物学评价这一环节中也将发挥至关重要的作用。
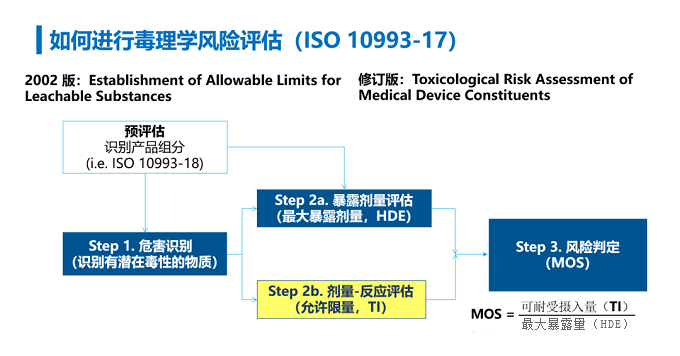
医疗器械的毒理学风险评估主要遵循ISO 10993-17。现行版本中描述了医疗器械可沥滤物的人体允许限量的确定方法,其最新修订草案中从整体框架上对产品中化学成分的毒理学风险评估进行了阐述,主要包括四个部分:危害识别、剂量-反应评估、暴露量评估、以及风险判定。即:识别器械中可能存在危害的物质,通过比较这些物质在人体中的无不良反应剂量与相应的人体暴露量,结合产品的临床使用情形、暴露风险、安全范围等,综合评估在临床使用过程中产品的可沥滤物对目标人群是否具有安全性风险。
具体评估思路如下图2:
医疗器械的生物学评价报告
综合上述生物学测试和评估结果的生物学评价报告是医疗器械产品注册申报的重要技术文件,用于确定所获取的信息是否已充分满足生物学安全性评价的目的并得出产品是否安全性的结论,属于产品风险管理的一部分。该报告通常包括以下几个方面的内容:
1)医疗器械生物学评价的策略和程序内容;
2)在风险管理计划范畴内确定材料可接受标准及预期用途;
3)充分的材料表征;
4)开展/豁免测试的说明;
5)已有数据和试验结果的解释;
6)完成生物学评价所需的补充数据;
7)器械总体生物学安全性的结论
